

Was man als BioinformatikerIn so wissen sollte – Teil 1
BLOG: Bierologie



Bioinformatik scheint immer noch eine beliebte Spezialisierung zu sein. Es gibt genug zu tun: Sequenziermaschinen (wie z.B. die neue PacBio RSII oder die Illumina Maschinen die das menschliche Genom endlich für weniger als 1000 Dollar auslesen können sollen) generieren mehr Daten für weniger Geld, und der Trend wird wohl auch nicht aufhören:
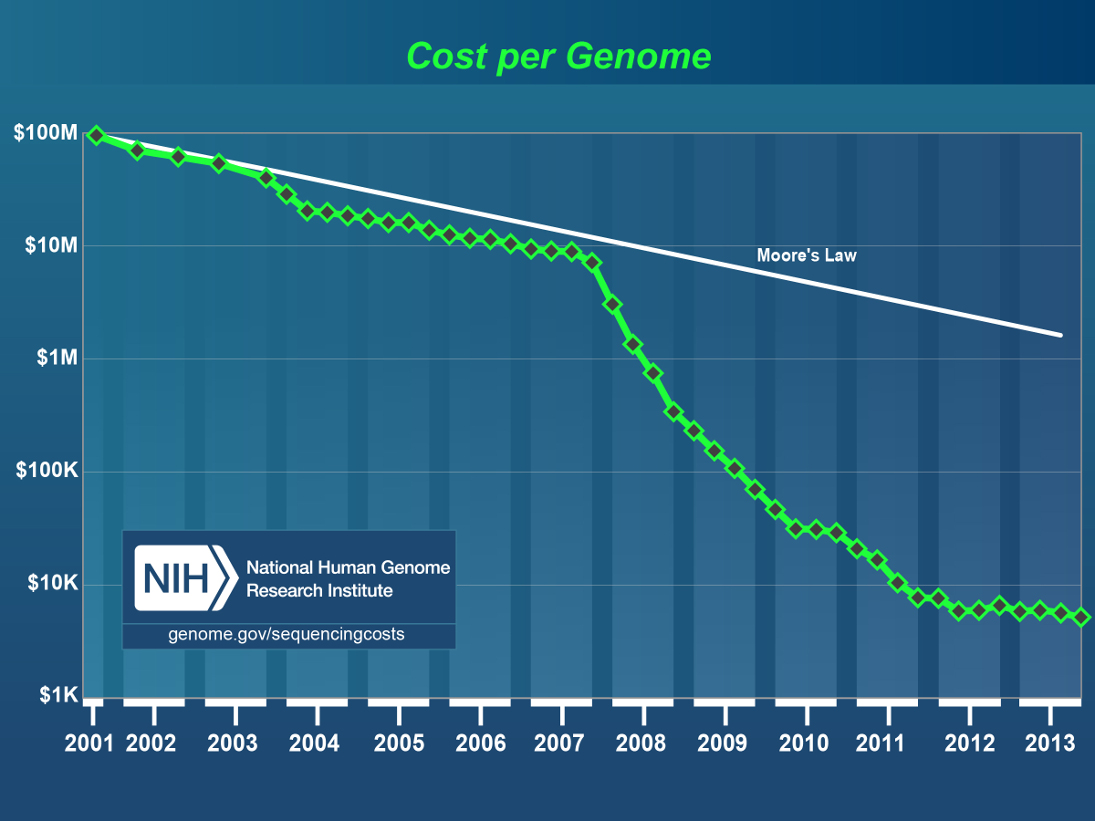
Kosten per sequenziertem Genom, von 2001 (ca. 100 Millionen US-Dollar) bis 2013 (ca. 7000 US-Dollar). Quelle.
Stellenangebote gibt es (für den wissenschaftlichen Standard) viele: NatureJobs listet momentan 497 Stellenangebote zum Stichwort “bioinformatics”, davon 40 in Deutschland. In Deutschland sieht studientechnisch die Lage gut aus – die 2008 erschienene Veröffentlichung A review of bioinformatics education in Germany beschrieb die damalige Situation in Deutschland. Bioinformatik Studiengänge gibt es in Deutschland seit 1998, mit steigender Tendenz: Wikipedia hat eine frei zugängliche Liste. Finanzierungstechnisch sah und sieht es in Deutschland gut aus.
Die Deutsche Forschungsgesellschaft begann 2000 mit der DFG-Initative Bioinformatik 5 Standorte mit jährlich 10 Millionen DM und das Bundesministerium für Bildung und Forschung (BMBF) fing an, 2001 “Bioinformatik Kompetenzzentren” mit 100 Millionen Mark zu fördern, und diie relativ gute Förderungssituation hält in Deutschland an, z.B: begann das BMBF 2013 mit der Einrichtung des “Deutschen Netzwerk für Bioinformatik-Infrastruktur”, einer Initative um insbesondere mit der riesigen Datenflut zurechtzukommen.
Die neue Veröffentlichung “Bioinformatics Curriculum Guidelines: Toward a Definition of Core Competencies” versucht, ein einheitliches Curriculum für Bioinformatikerinnen zu definieren. Dabei unterscheiden die Autoren zwischen drei “Untergruppen” – bioinformatics user, scientist und engineer. Ein/e bioinformatics engineer muss am meisten über die Implementation von Algorithmen Bescheid wissen und braucht weniger biologisches Fachwissen als ein bioinformatics scientist. Zusammen mit dem bioinformatics scientist wissen beide genug über ihre Disziplin, um Informatik-Fachwissen auf biologische Probleme anzuwenden; der/die scientist benutzt aber nur bereits entstehende Algorithmen, in bereits bestehenden und neuen Kontexten. Bioinformatics users sind 99.9% aller Biologen; die Wissenschaftlerinnen, die sich “nur” mit Biologie befassen und dann an ihrem PC die vorgefertigte Analyse durchführen, aber keine neuen Analysen erfinden oder Algorithmen unterschiedlich anwenden.
Wer sich wo einordnet, ist fließend – Philipp ordnet sich irgendwo zwischen bioinformatics engineer und scientist ein, Basti ebenfalls, aber mehr verstärkt mit einem Fokus auf scientist als engineer.
Dementsprechend ist es schwer, ein Kurrikulum zu definieren, dass alle Bereiche involviert. Dazu kommt erschwerend hinzu, dass unterschiedliche Spezialisierungen unterschiedliches Wissen brauchen. Wer Genome zusammenbaut muss über Graphentheorie Bescheid wissen, wer Protein-interaktion simuliert muss mehr über 3D-Strukturen wissen. Wer an was arbeitet stellt sich normalerweise nicht im Studium heraus; das entscheidet mehr oder weniger der Zufall der Doktorandenstelle, und welche Daten die Arbeitsgruppe so hat.
Für Leute die ohne formelles Kurrikulum in die Welt der Bioinformatik geschleudert werden gibt es von Nick Loman und Mick Watson in Nature Biotechnology einen netten Kommentar mit dem Titel So you want to be a computational biologist der generelle Ratschläge gibt, egal in welchen Bereich der Bioionformatik man eintauchen will.
Besonders freuen wir uns das die beiden Methodenverständnis und Tests hervorheben. Denn was im Labor eine Selbstverständlichkeit ist kommt bei der Arbeit am Rechner leider immer noch oft zu kurz. Dabei gilt dort das gleiche wie im regulären Labor: Wer nicht versteht was er tut hat keine Chance auftretende Probleme zu beseitigen oder erkennt im schlimmsten Fall nicht mal das die Ergebnisse keinen Sinn ergeben. Erst recht nicht wenn keine Positiv- und Negativkontrollen mitführt werden.
Ebenfalls dazu passend: Egal ob es um Daten oder Software geht, vertraue niemandem ist ein Motto was die angehende Bioinformatikerin im Kopf behalten sollte. Denn sonst stellt sich nach einigen verschwendeten Monaten raus das die Software die man benutzt hat doch nicht tat was sie angeblich erreichen sollte oder das der benutzte Datensatz gar nicht das beinhaltet was man dachte.
Den letzten wichtigen Tipp den Loman und Watson geben können wir beide ebenfalls nur vollumfänglich unterschreiben: Vernetzt euch, denn irgendjemand hat euer Problem vermutlich schon (fast) gelöst. Auf Diskussionsplattformen wie SEQanswers oder BioStars gibt es Leute die ähnliche Probleme schon mal gelöst haben und ihr Wissen gerne teilen. Und für uns beide war auch Twitter schon so manches mal bei der Problemlösung hilfreich.
Was wir als “Kernkompetenzen” der Bioinformatik ansehen kommt im nächsten Post!
Gibt es schon Ansätze aus den nunmehr verstärkt erfassten Datenlagen Information zu extrahieren?
MFG
Dr. W
Hallo,
an welche Informationen denken Sie genau?
Ein Beispiel ist k-mer counting:
Alle reads, die aus der Sequenziermaschine kommen, werden in “k-mers” aufgebrochen: überlappende Buchstabensequenzen als “vereinfachte” Repräsentation der Information im read. Simplifizierendes Beispiel wäre: ATGC wird in 2-mer aufgebrochen, damit bekommt man AT, TG und GC.
Es gibt inzwischen mehrere Programme die die k-mers zählen in einem kompletten Datenset zählen und damit allerhand anstellen:
– k-mers, die sehr selten auftreten, sind Fehler der Sequenziermaschine. Reads mit diesen k-mers kann man also kürzen oder wegschmeißen.
– manche Genom Assembler-Programmer funktionieren nicht gut, wenn sehr viel Daten eingelesen werden. khmer ist ein Programm, dass zu oft auftretende k-mers entfernt so dass alle k-mers ungefähr gleich oft auftreten – keine Information geht verloren, nur Daten.
– andere Fragen lassen sich beantworten: Wie sieht die Komposition des Genoms aus? Tauche manche Motive öfter auf als andere? Haben diese Motive biologische Funktion? etc.