

EVE: Eine Künstliche Intelligenz an der Schnittstelle zwischen Evolutionärer Medizin und Genommedizin
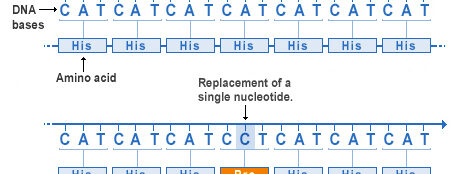
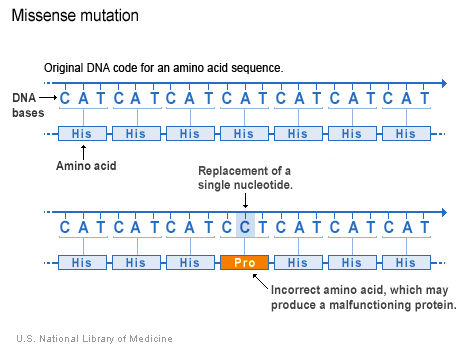
Die evolutionäre Medizin ist ein Zweig der Humanmedizin bei der Ärzt*innen im Stoffwechsel von Tieren nach neuen Therapieansätzen zur Behandlung menschlicher Krankheiten suchen. Ich möchte das am Beispiel einer Erbkrankheit der Leber erklären: Menschen, die an Progressive familiäre intrahepatische Cholestase Typ 3 (PFIC3) leiden, haben eine extrem niedrige Phospolipidkonzentration im Gallensaft, die mit erhöhtem Risiko für Gallensteine und Zerstörung der Gallenkanälchen verbunden ist. PFIC3 beginnt in der frühen Kindheit und macht durch ihren schweren Verlauf oft eine Lebertransplantation notwendig. Verantwortlich für PFIC3 sind Mutationen in den zwei, von Mutter und Vater geerbten, Abcb41-Genen, die dazu führen, dass diese Gene inaktiv sind und die Zellen kein Abcb4-Protein herstellen. Nur wenn das mütterliche und das väterliche Abcb4-Gen inaktiv sind kommt es zum Ausbruch von PFIC3.
Orthologe Gene und Erbkrankheiten
Das Abcb4-Gen codiert für ein Protein, das in der Leber hergestellt wird und von dort Phospholipide in den Gallensaft transportiert. Das Abcb4-Gen ist ein orthologes Gen. Orthologe Gene sind Gene, die in verschiedenen Organismen (z. B. Mensch, Maus, Fliege, Bäckerhefe, Sonnenblume) vorkommen und deren Nucleotidsequenzen eine hohe Übereinstimmung haben. Stammesgeschichtlich wird in diesem Fall ein gemeinsamer Vorfahre vermutet, aus dem heraus sich die entsprechenden Organismen entwickelt haben. Durch die hohe Übereinstimmung2 in der Nucleotidsequenz wird eine ähnliche Funktion der von ihnen codierten Proteine vermutet. Je näher Tierarten miteinander verwandt sind, desto höher ist die Übereinstimmung der Nucleotidsequenzen zwischen zwei orthologen Genen. Je näher Tierarten miteinander verwandt sind, desto mehr orthologe Gene haben sie gemeinsam. Da die Genetiker*innen in den letzten 24 Jahren3 nicht nur das Genom des Menschen, sondern auch die Genome von vielen Tieren sequenziert haben, sind bereits viele orthologe Gene entdeckt worden, die unter anderem in OrthoDisease dokumentiert werden. OrthoDisease ist eine Datenbank mit Genen von Modellorganismen, die ortholog zu menschlichen Genen sind, die Mutationen haben, die zu Erbkrankheiten führen. Diese tierischen Gene werden im Englischen Human Disease Orthologs genannt.
Das Abcb4-Gen ist im Meerschweinchen und im Pferd inaktiv, denn größere Teile der DNA-Sequenz fehlen in diesem Gen. Zusätzlich haben Meerschweinchen und Pferd extrem niedrige Phospolipidkonzentrationen im Gallensaft genau wie die Menschen, die an PFIC3 leiden. Es stellt sich also die Frage, warum diese zwei Säugetierspezies nicht an den für PFIC3 typischen Leberschäden leiden, obwohl das Gen inaktiv ist. Wie bleibt die Leber gesund? Um diese Fragen zu beantworten ist die Humanmedizin auf die Hilfe der Tiermedizin und der Bioinformatik angewiesen. Die Bioinformatik verfügt jetzt mit der künstlichen Intelligenz EVE (Evolutionary Model of Variant Effect) über ein neues Werkzeug sie dabei zu unterstützen.
Künstliche Neuronale Netzwerke und Unüberwachtes Maschinelles Lernen
EVE ist ein künstliches neuronales Netzwerk, das Unüberwachtes Maschinelles Lernen (UML) nutzt. UML beruht nicht auf vordefinierten Parametern und Regeln, sondern beinhaltet adaptives Lernen. Ein klassisches Beispiel ist das Lernen, was Katzen und Hunde sind von Katzen- und Hundebildern: Beim UML wird dem Netzwerk eine Reihe von Katzen- und Hundebildern vorgelegt, ohne dass ihm gesagt wird, welche Bilder Katzen zeigen und welche Hunde. Es muss das selbst erkennen. UML erkennt neue Muster in noch nie gesichteten Daten und das macht diesen Ansatz besonders geeignet für die Aufgabe.
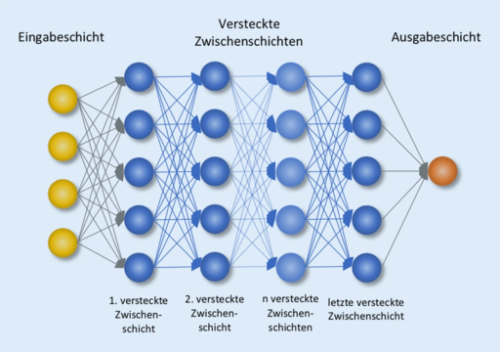
Die Forscher*innen Debora S. Marks (Medizinische Fakultät der Universität Harvard, USA) und Yarin Gal (Abteilung für Informatik der Universität Oxford, England) haben mit ihrem Team EVE entwickelt um Mutationen in den orthologen Genen von 140 000 Tierarten (inklusive ausgestorbener Tierarten) zu erkennen und zu klassifizieren. Die Forscher*innen bewerteten mit EVE 36 Millionen Proteinsequenzen und 3.219 krankheitsassoziierte Gene in verschiedenen Tierarten. EVE schätzte die Wahrscheinlichkeit, dass jede einzelne Aminosäurevariante entweder unbedenklich oder krankmachend für den Menschen ist.
Um festzustellen, ob EVE genaue Vorhersagen macht, verglichen die Forscher seine Ergebnisse mit fünf Mutationen in menschlichen Genen, deren klinische Bedeutung bekannt ist: verschiedene Formen von Krebs, mehrere Krebssyndrome und Herzrhythmusstörungen. Die Vorhersagen von EVE überschnitten sich mit den aktuellen Beschreibungen aus experimentellen Daten.
Ein bemerkenswerter Vorteil von EVE gegenüber den derzeitigen Methoden besteht darin, dass es einen kontinuierlichen Score statt eines binären Scores vergibt. Denn selbst wenn Genvarianten als krank machend eingestuft werden, ist die Art und Weise, wie sich eine Mutation im Körper auswirken kann, vielschichtig. “Es gibt ein ganzes Kontinuum der Pathogenität”, so Marks. “Der kontinuierliche Score ist sehr wichtig für die Vorhersage des Grades der Pathogenität. Bedeutet die Mutation, dass ich Schmerzen in meinem kleinen Zeh bekomme oder werde ich morgen sterben?” Die Forscher*innen müssen z. B. bedenken, dass eine Orthologie von Genen nicht zwangsläufig zu Orthologie von Organen führt, so ist z. B. der Fall nicht auszuschließen, dass orthologe Gene und damit auch orthologe Proteine in zwei völlig verschiedenen, nicht-orthologen Geweben gefunden werden.
Ein weiterer wichtiger Aspekt von EVE ist die Zuweisung eines Vorhersagesicherheitsscores für jedes einzelne Gen. Für jede genetische Variante teilt EVE den Ärzt*innen mit, wie sehr sie der Vorhersage vertrauen können. “Wir geben den Mediziner*innen nicht nur eine Zahl, sondern auch den Grad der Unsicherheit, der damit verbunden ist”, sagt Gal. “Das ist etwas, dass sie bei der Entscheidungsfindung verwenden können. EVE kann sagen: ‘Ich glaube, diese Variante gehört zu diesem Stapel, aber ich habe noch nie solche Varianten gesehen, also ist das mit Vorsicht zu genießen.
Von OrthoDisease zur Genommedizin
Marks und Gal beteiligen sich auch an der Atlas of Variant Effects Alliance, einer weltweiten Forschungsinitiative, deren Ziel es ist, einen umfassenden Atlas aller möglichen menschlichen Genvarianten und ihrer Auswirkungen auf Proteinfunktionen und Organe zu erstellen. Ziel der Bemühungen ist es, die Diagnose, Prognose und Behandlung menschlicher Krankheiten zu verbessern. Seitdem Next Generation Sequencing4 automatisierte Genomsequenzierungen im großen Maßstab möglich gemacht hat, verstehen Forscher*innen immer schneller, welchen Einfluss spezifische genetische Veränderungen auf die Gesundheit oder Erkrankung eines Menschen haben. Die Genommedizin nutzt Sequenzinformationen für eine genetische Diagnostik und klinische Interpretation der individuellen Erbinformation.
Fußnoten
1. Abcb4 ist die englische Abkürzung für ATP binding cassette subfamily B member 4
2. Stimmen zwei Gene in der Nukleotidsequenz in mehr als 30 % ihrer Nukleotide in der Abfolge überein, so gilt eine andere Ursache als die gemeinsame Abstammung als unwahrscheinlich.
3. 1998 wurde das Genom des Fadenwurms Caenorhabditis elegans sequenziert. Es war das erste Mal, dass das Genom eines Tieres sequenziert wurde. Das erste Genom eines Säugetieres war das des Menschen 2003.
4. Ein komplettes, menschliches Genom kann innerhalb eines Tages sequenziert werden.
Ja, was hier Joe Dramiga vorstellt, wird das Leben in 20 Jahren begleiten: Jeder/Jede wird wissen, was für ein Genom er/sie hat, Genom-Triplett für Genom-Triplett und ebenso wird das der Hausarzt wissen und dementsprechend die verordneten Medikamente wählen und Krankheitssymptome mit dem Wissen um die Gene seiner Patientin deuten. Ja, schon bei der Geburt wird das Gesamtgenom des Neugeborenen bestimmt werden, so dass man in Zukunft seltene Erbkrankheiten nicht mehr verpasst.
Jeder/jede von uns wir mit 100 Neumutationen geboren. Fast immer haben diese durch Zufälle im Erbmaterial der Eltern entstandenen Mutationen keine spürbaren Auswirkungen auf unser Leben. Wobei es gewagt ist und sehr relativ ist, zu sagen, nur wenige seien von seltenen vererbten Krankheiten betroffen, liest man doch auf der Website Seltene Erkrankungen des Bundesministeriums für Gesundheit (Zitat):
Fazit: Das hier von Joe Dramiga vorgestellte Geninterpretationssystem EVE gibt nicht nur Hilfestellung bei bereits bekannten Erbkrankheiten, sondern es schätzt das krankmachende Potenzial jeder Genvariante ein. Und wir wissen ja, dass jede/jeder von uns etwa 100 neu aufgetretene Genvarianten hat.
Im probabilistischen Sinne :
Sicherlich darf der Herr Allgemeinmediziner, hilfreich, auch die Genetik zK nehmen, um dann mit Medikation sozusagen noch ein wenig besser zu reichen.
Diesen “Sound”, Herr “Holzherr” (die doppelten Anführungszeichen nur deswegen, weil Sie ein als solches unerkennbares Pseudonym verwenden) im letzten Satz Ihrer zitierten Nachricht mag Dr. Webbaer dagegen nicht.
Ist das mit der Wahrscheinlichkeit auch bei Ihnen (vs. ‘Und wir wissen ja, dass jede/jeder von uns etwa 100 neu aufgetretene Genvarianten hat.’, danke für diesen Gag) verstanden worden?
Joe ist schon ganz OK.
Mit freundlichen Grüßen
Dr. Webbaer
Sicherlich liegt ein nutzenswerter Ansatz vor, der gerne auch durch bes. Berechnungen, gar durch AI vorgenommen, erfolgen darf bis soll :
Der Schreiber dieser Zeilen rät hier zum probabilistischen Denken, und dazu in Betracht zu ziehen, dass “gen- sequenzierte”, äh, Gene auch anders meinen könnten als krankheitserregend zu sein, Seiteneffekte meinend, die so, sozusagen statistisch nicht erkannt werden könnten, anzunehmenderweise so nicht erkannt werden könnten, die positiv sind und insofern nicht “auszumerzen”.
Dr. W mag so nicht :
-> https://de.wikipedia.org/wiki/CRISPR/Cas-Methode
Was auch damit zusammen hängt, dass die hier gemeinte Lebensdatenhaltung in etwa so-o groß ist :
-> https://de.wikipedia.org/wiki/Desoxyribonukleinsäure (‘Die Speicherkapazität der DNA ist extrem hoch und konnte bisher nicht technisch nachgebildet werden. Die in einem Teelöffel getrockneter DNA enthaltene Information entspricht einer Größenordnung von einer Billion Compact Discs zu je 650 Megabyte.’)
—
Sischer, bei Kartoffeln, Reis, Getreideprodukten generell darf hier geändert und auch vermampft werden.
Ansonsten darf gerne im probalistischen Sinne so versucht werden, auch wenn’s klar spekulativ bleibt.
Mit freundlichen Grüßen
Dr. Webbaer
PS:
Diesen “Sound” : ‘Seitdem Next Generation Sequencing automatisierte Genomsequenzierungen im großen Maßstab möglich gemacht hat, verstehen Forscher*innen immer schneller, welchen Einfluss spezifische genetische Veränderungen auf die Gesundheit oder Erkrankung eines Menschen haben. [Artikeltext]’ – mag Dr. Webbaer dagegen nicht so-o.
JFTR.