

Fatale Folgen für SFPQ bei Alzheimer
BLOG: Die Sankore Schriften

Bei Menschen mit Alzheimer bewirkt das Protein Tau in bestimmten Nervenzellen des Hippocampus, dass der Transkriptionsfaktor SFPQ aus dem Zellkern in das Zytoplasma transportiert wird. Dort kann er nicht mehr seine Funktion bei der Expression von bestimmten Genen ausüben.
Das Mikrotubuli-bindende Protein Tau und die Alzheimer-Krankheit
Das Tauprotein stabilisiert die Mikrotubuli, jene röhrenförmigen Proteinfasern, die ein wesentlicher Bestandteil des Grundgerüsts aller Zellen sind. In den Zellfortsätzen der Nervenzelle, den Axonen und Dendriten, läuft an ihnen wertvolle Fracht entlang – huckepack getragen von Motorproteinen wie dem Kinesin, einem zelleigenen „Frachtunternehmen“. Proteine, Nährstoffe, aber auch ganze Zellorganellen, wie Mitochondrien und Peroxisomen, kugelförmige Miniatur-Container für Enzyme. Tau bindet locker an die Mikrotubuli, stabilisiert sie und hilft so mit, dass der Transport in den Nervenzellen reibungslos läuft. Bei Alzheimer wird Tau von hyperaktiven Proteinkinasen wie beispielsweise der Kinase GSK3, überphosphoryliert. Das heißt, an vielen Stellen des Tauproteins sitzen nun plötzlich Phosphatgruppen. So kann es seine Stützfunktion nicht mehr ausüben. Die Tauproteine lösen sich von den Mikrotubuli ab und lagern sich zu unlöslichen, langen Fasern zusammen, den sogenannten Neurofibrillenbündel (im Englischen auch Neurofibrillary Tangles, NFT genannt). Diese Neurofibrillenbündel schaden den Nervenzellen dauerhaft, da sie nicht mehr abgebaut werden können.
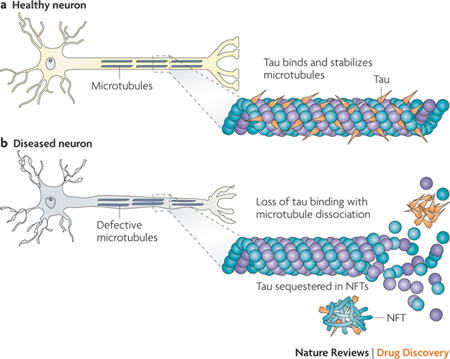
Abb.1: Die Bindung von Tau an Mikrotubuli in Nervenzellen
Das ist der Anfang vom Ende einer Nervenzelle; die Mikrotubuli gehen kaputt und damit die Schienen für den Transportverkehr. Die Motorproteine, die wie Züge arbeiten, können die Fracht nicht mehr transportieren. Dann verschwinden mangels Versorgung die Synapsen, jene winzigen Ausstülpungen der Axone und Dendriten, über die Nervenzellen chemisch und elektrisch miteinander kommunizieren. Daraufhin sterben Axone und Dendriten, und schließlich die ganze Nervenzelle. Diese spezifische Schädigung der Nervenzellen durch überphosphoryliertes Tauprotein findet man nicht nur bei Alzheimer, sondern auch bei einer Reihe anderer neurodegenerativen Erkrankungen des Gehirns wie z. B.: Pick-Krankheit, Frontotemporale Demenz und Parkinsonismus assoziert mit dem Mikrotubuli-assozierten Protein Tau (FTDP-MAPT), Kortikobasale Degeneration (KBD), Progressive supranukleäre Blickparese (PSB), die unter dem Namen Tauopathien zusammengefasst werden.
Umlagerung von Transkriptionsfaktoren vom Zellkern ins Zytoplasma bei neurodegenerativen Erkrankungen
Die von uns entdeckte Tau-vermittelte Fehllokalisation von SFPQ bei der Alzheimer-Krankheit und der Pick-Krankheit stellt eine bisher unbekannte Wirkung von Tau dar, dessen Konsequenzen für die kranken Nervenzellen nun weiter erforscht werden müssen [1]. Auffällig ist, dass in letzter Zeit mehrere Arbeitsgruppen, die über neurodegenerative Krankheiten forschen, über die Umlagerung von Transkriptionsfaktoren aus dem Zellkern ins Zytoplasma berichten [2]. So findet man bei Amyotrophe Lateralsklerose (ALS) und Frontotemporale Demenz (FTD), die Transkriptionsfaktoren TDP-43 und FUS im Zytoplasma [3, 4].
Die transgene pR5-Maus: Ein Mausmodell für FTDP-MAPT
Der Weg zu unserer Entdeckung führte über ein transgenes Mausmodell für die erbliche Form der Hirnkrankheit Frontotemporale Demenz und Parkinsonismus assoziert mit dem Mikrotubuli-assozierten Protein Tau. Bei der erblichen Form von FTDP-MAPT haben die betroffenen Patienten eine Mutation in dem Gen für das Tauprotein. 2001 hatte die Arbeitsgruppe von Jürgen Götz in Zürich den transgenen Mäusestamm pR5 geschaffen. Diese Mäuse besitzen zusätzlich das menschliche Tau-Gen mit der P301L-Mutation, die FTDP-MAPT auslöst. Durch die P301L-Mutation wird an der 301sten Aminosäure von Tau anstelle der Aminosäure Prolin die Aminosäure Leucin eingebaut. Dadurch entwickeln die Mäuse innerhalb von wenigen Monaten Alzheimer-ähnliche Symptome: Das Tau-Protein wird überphosphoryliert, es entwickeln sich Neurofibrillenbündel, die Nervenzellen werden geschädigt, und die Mäuse leiden unter Gedächtnisverlust.
Da sich die Neurofibrillenbündel zuerst im Mandelkern der pR5-Mäuse bilden, führten wir eine vergleichende Genexpressionsanalyse dieser Hirnregion bei den transgenen Mäusen im Vergleich zu normalen Mäusen (Wildtyp) durch, um herauszufinden wie das mutierte menschlich Tau die Genexpression dort beeinflusst. Dabei entdeckten wir unter anderem, dass das SFPQ-Gen in den pR5-Mäusen im Vergleich zu den Wildtypmäusen überexprimiert ist.
Umlagerung von SFPQ vom Kern ins Zytoplasma bei der Alzheimer-Krankheit und der Pick-Krankheit
Wir waren natürlich gespannt, wie sich SFPQ bei Tauopathien verhält, und schauten uns in Hirnschnitten von Alzheimer- und Pick-Kranken die intrazelluläre Lokalisation des SFPQ-Proteins an. Im Hippocampus, dem Sitz des räumlichen Gedächtnis, eine Region, die in Alzheimer besonders früh geschädigt wird, fanden wir diese zytoplamatische Fehllokalisation von SFPQ sowohl in Nervenzellen als auch in Astrozyten, den Stütz- und Nährzellen des Gehirns.
Die Braak-Stadien der Alzheimer-Krankheit
Die Neuropathologen Heiko und Eva Braak wiesen nach, dass die Lokalisation und Häufigkeit von Neurofibrillenbündeln präzise mit dem Fortschreiten der Alzheimer-Krankheit zusammenhängt. Ihre Einteilung der Krankheit in sechs Verlaufsstadien wird seit 1997 aufgrund einer Empfehlung des amerikanischen National Institute on Aging (NIA) international eingesetzt und ist zum “Goldstandard” der Alzheimer-Stadieneinteilung geworden.
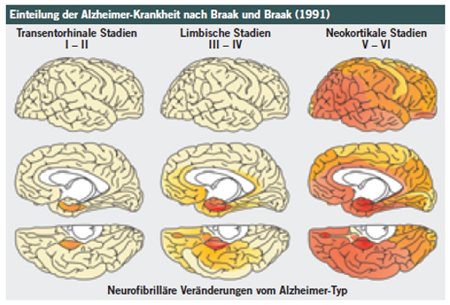
Abb.2: Die Braak-Stadien von Alzheimer Stadien I und II: Die Alzheimer-typischen Neurofibrillenbündel entstehen zunächst in Schläfenlappen des Gehirns, im transentorhinalen Kortex. Stadien III und IV: Weitere Teile des limbischen Systems, z. B. der Hippocampus sind von den Neurofibrillenbündel betroffen. Stadien V und VI: Die Neurofibrillenpathologie hat sich bei gleichzeitiger Zunahme der Intensität in den zunächst betroffenen Arealen weiter auf den Neokortex ausgedehnt; in diesen Stadien zeigen die meisten Patienten Symptome einer Alzheimer-Krankheit
Wir haben mit Hirnproben aus dem entorhinalen Kortex von Alzheimer-Kranken gezeigt, dass beim Durchgang durch die Braak-Stadien die Menge an SFPQ kontinuierlich abnimmt.
Tau-induzierte Umlagerung von SFPQ in der Zellkultur
In der Zellkultur konnten wir dann zeigen, dass eine Überexpression von Tau zu dieser Fehllokalisation von SFPQ führt. Wie das genau geschieht, soll durch weitere Experimente aufgeklärt werden.
Weiterführende Literatur
Excess Tau Protein Damages Brain’s GPS For Alzheimer’s Patients
Bildnachweis
Abb.1: Die Bindung von Tau an Mikrotubuli in Nervenzellen
Quelle: Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies
Kurt R. Brunden, John Q. Trojanowski & Virginia M.-Y. Lee
Nature Reviews Drug Discovery 8, 783-793 (October 2009)
FIGURE 2: Tau in healthy neurons and in tauopathies.
Abb.2: Die Braak-Stadien von Alzheimer
Quelle: Alzheimer Forschung Initiative e. V.
Pingback:Alzheimer
Pingback:SFPQ transportiert spezifische mRNAs vom Zellkern in das distale Axon › Die Sankore Schriften › SciLogs - Wissenschaftsblogs