

Wie vertrauenswürdig sind Proteindatenbanken?
BLOG: Bierologie

Mit Datenbanken hab ich im Rahmen meiner "Arbeit" viel zu tun gehabt – stets zur Annotation neuer, kurzer mRNA-Schnipselsequenzen (=ESTs), um mehr über ihre (mögliche) Funktion zu erfahren. Aber wie sehr kann man den Datenbanken und ihren funktionellen Annotationen vertrauen?
Es gab schon viele Studien zu dem Thema, aber diese 4 Tage alte Studie hat mich in der Höhe der ermittelten Fehlannotationen doch überrascht. Schnoes et al. haben sich dabei ein sehr gut (u.a. experimentell) annotiertes Set von Enzymfamilien genommen, um damit verschiedene Datenbanken (wie z.B. Swiss-Prot oder NCBI-NonRedundant) auf ihre Fehler und Lücken zu untersuchen.
Unterschiedliche Ergebnisse zwischen den untersuchten Datenbanken sind unvermeidlich – Swiss-Prot z.B. wird komplett manuell annotiert, also würde man dort die geringste Fehleranfälligkeit erwarten. NCBI-NR dagegen ist meiner Erfahrung nach ein Riesenberg an Daten – so sind alle öffentlich erhältlichen Proteinsequenzen in dieser Datenbank enthalten, was für manuelle Bearbeitung einfach zu viel ist. Bei meiner Arbeit haben sich die Ergebnisse aus NCBI-NR meist auch als unzuverlässig erwiesen.
Aber wie sehen die genauen Ergebnisse der Forscher aus?
Als Testset dienten Enzyme aus 37 Superfamilien aus der SFLD-Datenbank, einer sehr gut (auch manuell) annotierten, in der alle Ergebnisse auch experimentell unterstützt worden. (Kurze Erklärung: Superfamilien fassen Familien mit ähnlichen Aufgaben zusammen)
Wie erwartet, liegt die Fehlerhöhe bei Swiss-Prot bei fast 0% – hier kann man den Ergebnissen also fast uneingeschränkt vertrauen. Bei NCBI-NR siehts ganz anders aus – mit den Jahren schleichen sich nur mehr und mehr falsche Einträge ein, wie man aus folgender Grafik entnehmen kann:
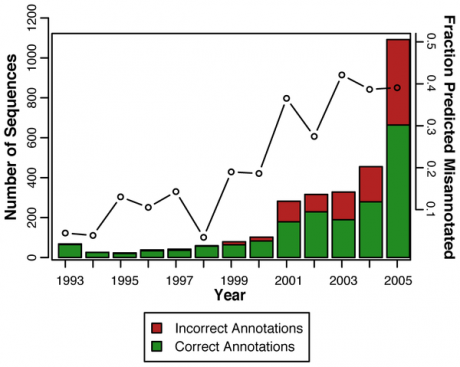
Quelle: Aus der Originalveröffentlichung
Die schwarzen Punkte stehen für den prozentualen Anteil an hinzugefügten falsch annotierten Sequenzen. Wie man sieht, wurden in manchen Jahren bis zu 40% falsche Annotationen hochgeladen! Die Diskrepanz lässt sich nicht mit einer erhöhten Anzahl von hochgeladenen Sequenzen erklären, da ja auch der prozentuale Anteil von falsch annotierten Sequenzen ansteigt. Anscheinend wurden die Forscher mit der Zeit nur schlampiger, oder sie verlassen sich zu sehr auf die Hilfe von bereits annotierten Sequenzen. Bei solchen Zahlen kann man sich doch auf keine Ergebnisse aus NCBI-NR verlassen. In der HAD Superfamilie war die durchschnittliche Misannotationsrate (in den Datenbanken TrEMBL, NCBI-NR und KEGG) sogar bei 60%!
Woher aber kamen die Fehler? In der Analyse haben sich 4 grundlegende Typen von Fehlen herausgestellt: bei 51% aller Fehler hat sich herausgestellt, dass das untersuchte Protein der falschen, aber zur richtigen sehr ähnlichen Familie zugeordnet wurde. In diesen Fällen bekommt man als Endbenutzer der Datenbank also schonmal eine recht gute Vorstellung von der Aufgabe des zu untersuchenden Proteins, als (guter?) Forscher kann man den Ergebnissen aber immer noch nicht zu 100% vertrauen. Bei 31% haben die Datenbanken "zuwenig" vorhergesagt – in diesen Fällen wurden die Proteinen den generelleren Superfamilien zugeordnet, aber nicht den (Unter-)familien, zu denen sie eigentlich gehören. In
9% konnten die Enzyme nicht den jeweiligen Superfamilien zugeordnet werden, und in den restlichen 6% wurden die Enzyme zwar der richtigen Familie zugeordnet, aufgrund eines Austauschs einer Aminosäure o.ä. in der zu untersuchenden Sequenz konnten sie von der untersuchen Datenbank aber nicht genauer zugeordnet werden.
Was bedeutet dass für den Forscher? Glaubt nicht 100%-ig euren Annotationsergebnissen (auch nich denen aus Swiss-Prot!), nehmt die Ergebnisse lieber als Anhaltspunkt für die mögliche Aufgabe eures Proteins. Für die Nicht-Forscher -fällt euch was ein? Also mir nicht.
Naja, eine falsche Zuordnung zu einer Familie ist gar nicht mal sooo tragisch. Mich nerven da eher die Einträge, die eine falsche Sequenz haben. Beispielsweise etwas das angeblich aus dem menschlichen Genom stammt, aber durch einfaches Blasten als bakterielle Vektorsequenz identifizierbar ist. Oder auch ganz gern mal total falsch vorhergesagte Spleißvarianten.
Aber schon lustig, als ich den Titel gelesen hab, dachte ich erst an die gefälschten Strukturdaten in der Protein Data Bank (z.B. bei WeiterGen oder im Nature Network).
Ja, komplett falsche Sequenzen hatte ich auch schon tausendmal 🙂 Das kommt davon wenn man nicht Software wie cross_match oder pregap4 benutzt um die ganzen Vektor- und Adaptersequenzen rauszuhaun (was wesentlich frickeliger ist als es sich anhört!).
Bioinformatik ist manchmal Magie, schwärzer als jede Proteinpurifizierung.