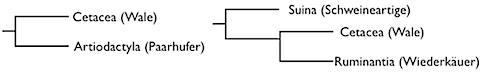
Darüber wie man mit Hilfe von DNA- und Protein-Sequenzen versuchen kann, den Stammbaum des Lebens aufzudröseln haben wir bei der Bierologie schon so manches Posting geschrieben. Und vermutlich in jedem der Postings haben wir auch auf die Fallstricke, die solche Methoden haben, hingewiesen. Da wären Unterschiede in der Rate, mit denen Substitutionen sich ansammeln, Rückmutationen, die dazu führen, dass man die ganzen Substitutionsraten unterschätzt und noch vieles mehr. Trotzdem kann man damit tolle Dinge rausfinden bzw. bestätigen. Es ist zum Beispiel noch gar nicht so lange her, dass man den Walen (Cetacea) mit Hilfe von molekularen Daten ihr Plätzchen im Tree of Life gegeben hat. Während die Analyse von morphologischen Daten schon etwas länger dafür sprach, dass sie mit den Paarhufern (Artiodactyla), also mit Schweinen, Kühen, Hirsche und Co. nah verwandt sind, sprechen die molekularen Daten dafür, dass die Wale in die Gruppe der Paarhufer gehören.
Danach sind die alle Schweineartigen abgespalten, bevor sich die restlichen Artiodactyla in Wale und Wiederkäuer aufgespalten haben. Durch die molekularen Analysen hat man darauf hin dann auch die Übergruppe der
Cetartiodactyla geschaffen (bislang hat diese Gruppierung noch keine taxonomische Einheit zugewiesen bekommen, so weit ich weiss). Anstatt sich aber nur DNA- oder Protein-Sequenzen anzuschauen, kann man auch andere, molekulare Marker benutzen, um Aussagen über Stammbäume treffen zu können. Im letzten Modul meines Studiums haben wir uns deshalb auch mit sogenannten
SINEs, den
short interspersed nuclear elements beschäftigt. Das sind kurze (100 bis 400 Basenpaare lange) Elemente, die man in den Genomen von sämtlichen Säugetieren finden kann und die zu den
Retroposons, die ihrerseits wieder zu den
Transposons gehören.
So weit verwirrt? So gut. Die Transposons werden umgangssprachlich auch gerne als „springende Sequenzen“ bezeichnet, da sie ihre Position im Genom ändern können. Das geschieht entweder per cut & paste, dann findet man das Transposon nach einer solchen Transposition einfach an einer anderen Stelle im Genom, oder per copy & paste. Dann findet man die Sequenz nach der Transposition an 2 Stellen. Die Retroposons nutzen dabei für ihre „Sprünge“ eine RNA-Zwischenstufe. Die SINEs, von denen es viele verschiedene Typen (abhängig von den Familien in denen sie vorkommen) gibt, nutzen also auch eine RNA-Zwischenstufe und, erfreulicherweise, springen sie nicht per cut & paste, sondern per copy & paste.
Und genau das kann man ausnutzen, um sich die Verwandtschaftsverhältnisse von Arten anzuschauen. Denn wenn ein
SINE erstmal an eine Stelle gesprungen ist, dann bleibt es mit hoher Wahrscheinlichkeit auch dort. Und es ist gleichzeitig unwahrscheinlich, dass es 2x unabhängig voneinander an die gleiche Stelle springt. Wenn man also 2 nah verwandte Arten untersucht, und dabei feststellt, dass beide Arten an einer Stelle ein
SINE besitzen, dann kann man davon ausgehen, dass bereits der letzte gemeinsame Vorfahr diesen
SINE an der Stelle besessen hat. Die entsprechenden Unterschiede kann man sogar schon sehen, wenn man die entsprechenden Stellen des Genoms amplifiziert und dann mit einer
Gelelektrophorese auftrennt.
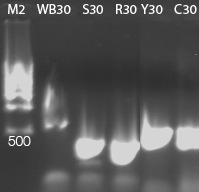
Bei einer solchen
Gelelektrophorese werden die vervielfältigten DNA-Stücke nach ihrer Länge aufgetrennt. Je weiter unten die Stücke im Foto sind, desto weiter sind sie auf dem Gel gereist. Und je weiter sie gereist sind, desto kleiner sind sie. In der ganz linken Spur sieht man den Marker, zum Größenvergleich. Die unterste, markierte Bande ist 500 Basenpaare groß. Die Bande darüber ist 1000 Basenpaare groß.
Und in diesem Bild sieht sieht man auch schön, dass die
Säbelantilope (S30) und das
Reh (R30) kleinere DNA-Fragmente, von knapp unter 500 Basenpaaren länge, zeigen, als
Wasserbüffel (WB30),
Yak (Y30) und
Kuh (C30), deren Fragmente etwas über 500 Basenpaare lang sind. Sprich: Während Wasserbüffel, Yak und Kuh ein
SINE an der betrachteten Stelle zeigen, haben Reh und die Säbelantilope keins. Das spricht stark dafür, dass bereits der gemeinsame Vorfahr von Wasserbüffel, Yak und Kuh diesen
SINE hatte, während sowohl die Säbelantilope als auch das Reh sich bereits vorher von dieser Linie abgespalten haben.
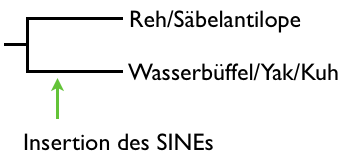
Das ist für euch vielleicht nicht so überraschend und stimmt auch mit den bekannten Stammbäumen so überein, aber es zeigt, wie die Methode an sich funktioniert. Und gleichzeitig ist die Aussagekraft so natürlich auch recht begrenzt, immerhin kann man so nur in 2 Gruppen einteilen: Diejenigen die es haben und diejenigen, die es nicht haben. Spannender wird es dann, wenn man eine ganze Reihe an verschiedenen SINEs betrachten kann. Hätten wir beispielsweise noch einen der nur in Yak und Kuh vorkommen würde, dann wäre das ein Zeichen dafür, dass der Wasserbüffel sich vor der Aufspaltung von Kuh und Yak aus der Linie abgespalten hat.
Nichtsdestotrotz gibt es auch bei dieser Methode ein paar Probleme: Zum einen muss es nicht zwangsläufig so sein, dass gleich große DNA-Fragmente auch wirklich das gleiche Element enthalten. Es können ja auch irgendwann im Laufe der Jahre unterschiedliche SINEs an die Stelle gesprungen sein. Aber die Fälle lassen sich noch durch eine einfache Sequenzierung und einen anschliessenden Sequenzvergleich erkennen (den man deshalb immer machen sollte). Problematischer ist es da schon, wenn ein SINE 2 mal unabhängig voneinander an die gleiche Stelle gesprungen ist, da hilft dann so einfach auch kein Sequenzvergleich mehr. Das ist zwar unwahrscheinlich, aber nicht ausgeschlossen. Aber da hilft es dann, wenn man eine größere Gruppe von Tieren vergleicht und mehr sich mehr als einen SINE anschaut.
Damit hat man neben den morphologischen Analysen und den sequenzvergleichenden, molekularen Methoden, jetzt auch noch ein anderes Werkzeug dafür, um seine Stammbäume weiter aufzulösen. Ich bin gespannt, welche Tiere dann als Nächstes neu einsortiert werden dürfen.



Schöne Erklärung, Danke! Was kann man denn sonst noch so für Elemente benutzen, um Tiere in den Tree of Life einordnen zu können? Da hast du als Bioinformatiker sicher viel mehr Ahnung als ich! Die Qualität der Gelelektrophorese lässt allerdings zu wünschen übrig, scheint was schief gelaufen zu sein. Ist das direkt aus dem Paper?
Elemente
Ich hab auch nicht so richtig den Überblick, welche Elemente man noch verwenden kann. Aber ich vermute, dass man auch LINEs und andere Transposon-Elemente verwenden kann, solange sie sich per Copy & Paste anstatt durch Cut & Paste durch das Genom fortpflanzen.
Ich weiss nicht, wie es mit Dingen wie CNVs aussieht, also ob es da auch welche gibt die phylogenetisch aussagekräftig sind, weiß ich nicht.
Zu dem Gelfoto: Das ist direkt aus unserem Praktikum, nicht aus einer echten Publikation 😉