Im letzten Beitrag hab ich ja angekündigt, dass ich mich mit den Ergebnissen und der Aussagekraft meiner Genotypisierung noch etwas näher auseinandersetzen will. Und über das Wochenende hab ich auch ein bisschen Zeit dafür gefunden. Da so ein Wochenende dann aber doch etwas kurz ist, um gut 1 Million Single Nucleotide Polymorphisms (SNPs) per Hand abzugrasen, von der fehlenden Lust dazu mal ganz abgesehen, hab ich mir ein kleines Script geschrieben, dass für mich die wirklich gut gepflegte SNPedia abgrast (das gibt es, falls Interesse besteht auch ebenfalls bei GitHub zum Download). In der SNPedia sind zur Zeit fast 23000 SNPs aufgelistet. Für viele dieser SNPs gibt es auch eine Übersicht über die verschiedenen Genotypen bzw. Varianten, die an der Stelle vorhanden sein können. Und für einen guten Teil der Einträge gibt es auch weitere Informationen darüber, welche Auswirkungen die einzelnen Genotypen des SNPs an der Stelle haben.
Ebenfalls im letzten Posting hatte ich schon überlegt, mir doch mal anzusehen, was für Informationen über seine nächsten Verwandten aus dem Datensatz so rausbekommen kann. Ich hab mich dann aber doch erstmal gegen eine Analyse des Y-Chromosoms und des Mitochondriums entschieden und einen etwas anderen Ansatz gewählt. Aber dafür steigen wir jetzt noch einmal kurz in die Mendelsche Genetik ein. Anstatt mit Erbsen machen wir das ganze direkt mal etwas abstrakter und schauen uns 2 theoretische Allele B und b an. Die Grafik hier unten zeigt das Punnett-Quadrat der möglichen Genotype und 2 mögliche Varianten an.
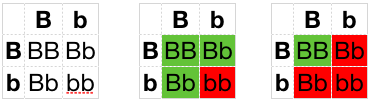
Das linke Quadrat zeigt uns einfach die möglichen Kombinationen, die in diploiden Organismen, wie uns Menschen z.B., entstehen können, wenn es 2 verschiedene Allele gibt. Nehmen wir nun an, dass B dafür sorgt, dass unser theoretischer Organismus Grün wird und b sorgt dafür, dass er Rot wird. Der mittlere Fall zeigt was passiert, wenn B dominant ist: Die Organismen die nur B tragen werden genauso Grün, wie jene die sowohl B als auch b tragen. Der recht Fall zeigt das Gegenteil: B ist rezessiv, b ist dominant. Und wer nur ein B hat, der bleibt Rot.
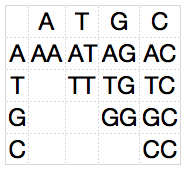
Das gleiche Spielchen kann man, wie in der Grafik hier oben gezeigt (der Übersichtlichkeit hab ich die Dopplungen einfach mal rausgelassen. Da die Tabelle an der Diagonalen gespiegelt ist, macht das nichts), auch für die 4 DNA-Basen durchführen. Und anstatt hier nur wieder mit ausgedachten Beispielen zu arbeiten, schauen wir uns einmal exemplarisch den SNP mit dem Namen
Rs9332964 an. Für diese Position sind laut Aussage der
SNPedia drei verschiedene Genotypen bekannt: AA, AG und GG. Während die beiden Genotypen AG und GG unauffällig sind, sind männliche
Homo sapiens, die den Genotyp AA tragen, betroffen. Und zwar von einem
Mikropenis. Sind nun alle wieder wach? Gut, dann können wir ja weiter Theorie machen:
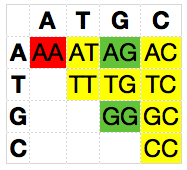
Wenn man die bekannten Genotypen nun in unser Punnett-Quadrat einträgt, dann ergibt sich folgendes Bild: Rot markiert ist der Mikropenis-Genotyp, Grün markiert ist der Genotyp für kein Mikropenis, in Gelb markiert sind die Genotypen für die in der SNPedia keine Daten vorliegen. Gegenüber dem Allel G ist das Mikropenis-Allel A also rezessiv. Dummerweise hat man hier leider noch keine Informationen darüber, ob A eventuell dominant gegenüber T oder C wäre, aber zu dem Problem kommen wir später noch einmal. Gehen wir der Einfachheit erstmal davon aus, dass A in diesem Beispiel komplett rezessiv ist, also auch die Genotypen AT und AC erfreulicherweise keinen Mikropenis haben und auch weder T noch C in irgendeiner Form mit Mikropenes zu tun haben (also auch z.B. TG oder CG keinen Mikropenis hervorrufen).
In diesem Fall kann man nämlich sehr einfach Aussagen über das Mikropenis-Risiko der nächsten Verwandten treffen. Wer selbst den Genotyp GG hat, der wird seinen Kindern auf jeden Fall ein G vererben. Damit werden sie auf jeden Fall keinen Mikropenis erben. Und da beide Elternteile ebenfalls mindestens ein G-Allel tragen müssen, wenn man selbst den Genotyp GG hat kann man auch sagen, dass weder Mama noch Papa einen Mikropenis haben. Gut, Mama dürfte das unmittelbar vermutlich so oder so weniger betreffen und auch bei Papa könnte man das auch ganz ohne SNP-Analyse noch einfach rausbekommen.
Spannender wird es dann, wenn man sich SNPs anschaut, die mit Krankheiten assoziiert sind, die man nicht so einfach erkennen kann. Krebs ist dafür z.B. ein ganz gutes Beispiel. Der SNP
Rs7837688 z.B. ist mit einem gesteigerten Prostatakrebs-Risiko assoziiert. Die Genotypen GG und GT haben gegenüber dem Genotyp TT ein um 1,7 fach erhöhtes Prostatakrebs-Risiko. Platt gesagt: Das Krebs-Allel G ist dominant gegenüber dem Allel T.
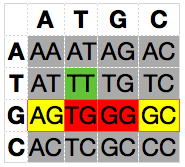
Und damit kommen wir mal wieder kurz zur Verwandtschafts-Analyse: Ich selbst habe den Genotyp GG, also müssen meine Eltern beide mindestens ein G tragen, sie fallen also in die dritte, rot umrandete, Zeile. Sollte Allel G auch in den anderen beiden Genotypen (hier mangels Wissen wieder in Gelb markiert, ausgegraut sind die Genotypen, die man hier gerade nicht betrachten will) dominant sein, dann wäre klar, dass sie auch direkt betroffen sind. Und genau diese Art von Vergleich hab ich am Wochenende mal angestellt. Dazu hab ich meine gesamten SNPs, an denen ich homozygot bin (also 2 gleiche Allele trage), mit der Datenbasis der SNPedia verglichen und geschaut, welche davon überhaupt gelistet sind.
Nach diesem ersten Schritt der Datenfilterung blieben bei mir noch 8600 SNPs übrig. Für diese 8600 habe ich dann geschaut, ob die SNPedia auch Genotypen an den Positionen kennt. Ergebnis: Es bleiben 8410 SNPs übrig. Im letzten Schritt habe ich dann geschaut, für welche Genotypen überhaupt ausreichend Beschreibungen vorhanden sind (welche Auswirkungen hat es, wenn man Genotyp X ist?). Das Ergebnis: Es gibt für meinen Datensatz 2245 ausreichend beschriebene Genotypen, die sich auf 753 meiner SNPs verteilen. Und in gut 120 Fällen ist das Allel, dass ich homozygot trage, dominant gegenüber dem Vergleichs-Genotyp aus den SNPedia-Daten.
Das klingt jetzt vielleicht gar nicht so schlecht, ist aber doch ernüchternd: Denn es macht im Schnitt etwas unter 3 Genotypen pro SNP. In jedem der 753 SNPs, die ich betrachtet habe, hat man also im besten Fall das gleiche Bild wie in den Beispielen Mikropenis oder Prostatakrebs: 50 % der möglichen Eltern-Genotypen sind nicht bekannt bzw. nicht beschrieben. Oder kommen sie einfach generell nicht vor bzw. sind so extrem selten und sind deshalb nicht verzeichnet?
Wenn sie so selten wären bzw. nicht vorkommen würden, dann wäre es egal, dass sie nicht verzeichnet sind: Dann könnte man mit ausreichender Sicherheit sagen, dass der elterliche Phänotyp meinem gleicht. Wären sie einfach aus anderen Gründen nicht beschrieben, dann könnte die Aussagekraft meiner Genotypen rapide sinken: Mit 50% Wahrscheinlichkeit sagen sie den elterlichen Phänotyp (in)korrekt vorher.
Die Analyse der Daten ist also nicht so einfach, wie man das vielleicht denken könnte. Mein Problem dabei: Ich bin kein Humangenetiker und auch kein Experte für die Analyse von Single Nucleotide Polymorphisms. Die Grundidee ist mir Ende letzter Woche relativ spontan gekommen und der Programmcode, um die Datenbanken abzufragen, wurde genauso spontan zusammengekritzelt. Falls ihr also gute Ideen habt, wie ich diese Unsicherheit des Fehlers ausmerzen kann: Nur zu, meldet euch in den Kommentaren. Vielleicht ist die Grundidee ja auch schon total bescheuert, oft steht man ja so auf dem Schlauch.
Meine Idee zum weiteren Vorgehen bislang: Es gibt ja erfreulicherweise schon einige andere Menschen, die ihre Typisierungs-Rohdaten veröffentlicht haben. Die werde ich mir mal besorgen und danach scannen, welche Genotypen sie an meinen Kandidaten-SNPs haben. Wenn die Anzahl der durchsuchten Typisierungen hoch genug ist, und man neben den bereits in der SNPedia gelisteten Genotypen keine anderen finden kann, dann spricht es auf jeden Fall dafür, dass alternative Genotypen zumindest selten sind.




Du kannst das Punnett-Quadrat meiner Meinung nach nicht wirklich auf SNPs anwenden, wie du es hier ja gemacht hast. Aufgrund des genetischen Codes und der Wobble-Hypothese, kann ein SNP nämlich mehrere unterschiedliche Folgen haben, was du mit dem Punnett-Quadrat so nicht erfassen kannst. Du müsstest also erst einmal schauen, wann dein SNP eine andere Aminosäure kodiert und wann nicht. Das hast du völlig vernachlässigt, ich denke aber, dass das sehr wichtig ist, weil eben veränderte Aminosäuren und so Proteine ja für ein erhöhtes Risiko verantwortlich sind. Ich weiß auch nicht ganz, wie ich die hier erwähnten dominanten und rezessiven Genotypen einstufen soll. Soll dominant bedeuten, das eine andere Aminosäure kodiert wird und rezessiv, dass das Codon zwar verändert wird, die Aminosäure aber gleich bleibt? Ich glaube hier fehlt eine Definiton, wann ein SNP dominant ist und wann nicht und was es schlussendlich bedeutet.
Punnett-Squares
Wir hatten ja gerade schon kurz auf Twitter darüber diskutiert, aber für die anderen Leser hier noch einmal:
Natürlich wäre es der Idealfall, wenn jeder Genotyp an einem SNP klar für ein synonyme oder eben nicht synonyme Mutation, also eine oder keine Änderung einer Aminosäure hervorrufen würde. Und so Fälle gibt es ja auch: Da ändert sich eine Base, damit eine Aminosäure, damit die Struktur des Proteins. Und schon geht etwas nicht mehr oder auf jeden Fall anders als ohne diese Mutation. Etwas ähnliches kann natürlich auch in Kontroll-Regionen passieren, so das man weniger/mehr Proteine erzeugt oder so etwas. Und auch auf der RNAi-Ebene kann man so etwas erzeugen.
Leider gibt es solche Daten über wirklich konkrete Veränderungen nur für relativ wenige SNPs und das ist auch meistens überhaupt nicht das, was man beim SNP-Mining macht. Stattdessen veranstaltet man Dinge wie Genome Wide Association Studies (GWAS) bei denen man einfach schaut, welche SNPs besonders häufig mit Krankheiten zusammen vorkommen.
Dann kann man sagen: “Wer Genotyp AA hat, der hat in 95 % der Fälle auch Krankheit X”, weiss allerdings genau gar nichts darüber, wieso das so ist. Es kann nämlich auch einfach so sein, dass die Auslösenden Mutationen gar nicht der SNP selbst sind, sondern nur in der Nähe sind. Über Linkage Disequilibrium werden SNP-Genotyp und Krankheit dann meistens zusammen vererbt.
Deshalb mein vereinfachtes Modell der Genotypen wie bei Mendels Grundlagen. Ich denke, dass man das durchaus vergleichen kann. Es gibt ja nicht immer nur 2 Allele an eine Ort. Nehmen wir als vergleichbaren, bekannten Fall nur mal das AB0-Blutgruppen-System (mir ist klar, dass auch das genetisch stark vereinfacht ist).
Aber dort gibt es als Haupttypen A, B und 0. Um es mit DNA vergleichbar zu machen, müsste man 4 Varianten haben. Splitten wir also A der Einfachheit halber einfach mal in die 2 bekanntesten Subgruppen A1 und A2 auf.
A1 ist afaik dominant über A2. A1, A2 und B sind dominant über 0. A1, A2, B sind co-dominant. Die 4 verschiedenen Varianten könnte man dann imho genotypisch genauso auf DNA-Ebene überführen, theoretisch könnten die 4 Blutgruppen-Dings dann sogar an einem SNP ablesbar werden. 🙂
Nur die Blutgruppen wären, mangels vollständiger Dominanz eines Allels, eben einer der SNPs mit denen ich in meiner Auswertung nichts anfangen kann. 🙂