

Von Transkriptomen und Genomen
BLOG: Bierologie

In meiner Bachelor-Arbeit habe ich mich ja mit der Analyse von Transkriptom-Daten beschäftigt. Wer schon wieder völlig vergessen hat was ein Transkriptom ist und wie man damit arbeiten kann, der sollte vielleicht noch einmal die groben Fakten dazu in diesem Blogpost nachlesen. Mittlerweile arbeite ich ja mit genomischen Daten, und dabei sind die Probleme dann auf einmal ganz andere. Denn so ein Genom besteht zum einen aus einer viel größeren Menge DNA als ein Transkriptom. Bei uns Menschen sind es z.B. nur 3 Prozent des Genoms, die für protein-codierende Sequenzen stehen. Und so das Transkriptom ausmachen. Nimmt man nun die restlichen 97 Prozent dazu wächst die Datenmenge und damit auch der Aufwand für Berechnungen und Analysen natürlich rapide an. Das ist aber noch gar nicht das Hauptproblem. Denn wenn der Computer länger zum berechnen braucht mag das ärgerlich sein, aber im Zweifel noch zu verschmerzen.
Problematischer ist dabei, wie Genome – zumindest von Eukaryoten – organisiert sind. Bei den Prokaryoten ist es, genau wie bei Transkriptomen, ganz einfach Gene zu finden. Diese starten immer mit der DNA-Sequenz ATG, dem sogenannten Start-Codon, und hören mit einem Stop-Codon (Der Sequenz TAA, TAG oder TGA) auf. Im Endeffekt muss man die möglichen Leseraster einer transkriptomischen oder auch einer prokaryotischen DNA-Sequenz nur auf diese Markierungen hin abscannen und hat dann potentielle Gene gefunden. Im Idealfall kann man diese, so gefundenen, Kandidaten-Sequenzen dann einfach auf eine der Sequenzdatenbanken werfen und schauen ob es schon bekannte Sequenzen mit bekannter Gen-Funktion aus diesem, oder einem nah verwandten, Organismus gibt.
Bei den Genomen der Eukaryoten funktioniert das nicht so einfach. Zwar starten Gene hier genauso mit dem Start-Codon und enden mit einem der Stop-Codons, es gibt aber ein großes aber: Denn die codierenden Bereiche werden von den nicht-kodierenden Introns in Exons aufgespalten. So, ist die Verwirrung jetzt komplett? Dann schauen wir uns einfach mal diese Grafik an:
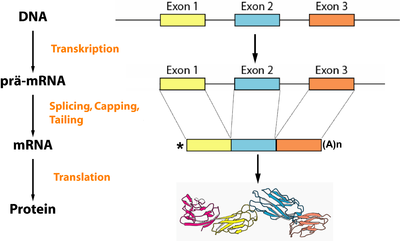
Mit der DNA ist in der Grafik ein Teil des Genoms, also ein Gen, beschrieben. Dieses Gen besteht auf der genomischen DNA noch aus 3 Exonen. Im Zuge der Transkription entsteht erstmal eine vorläufige RNA, in dieser befinden sich auch weiterhin noch die Introns. Erst danach werden diese Introns herausgespleist, und es passiert noch etwas Kosmetik um die Translation zum fertigen Protein zu ermöglichen. So weit, so gut. Dummerweise können sich funktionslose Start- & Stop-Codons auch in den Introns verstecken. Da die Introns ja sowieso weggeschmissen werden ist es biologisch kein Problem wenn solche Codons da drin stecken. Für die Erkennung der Gen-Sequenz ist das aber sehr wohl ein Problem. Denn der einfache und "dumme" Ansatz nach den Codons zu schauen funktioniert damit nicht mehr.
Um dieses Problem zu lösen gibt es diverse Software-Ansätze. Die meisten basieren dabei auf einem statistischen Modell, auf sogenannten Hidden Markov Models (HMMs). Grob gesagt versucht man dabei aufgrund von Beobachtungen Rückschlüsse auf einen versteckten Grundstatus zu ziehen. Damit solche Modelle allerdings gut funktionieren muss man die einzelnen Parameter des Modells erstmal herausfinden oder antrainieren. Und da die Verteilung und Erkennung von Introns dazu auch noch von der Art des Organismus abhängt den man untersuchen will, muss man das Training der Modelle eigentlich jedes Mal auf ein Neues machen. Im besten Fall hat man neben dem Genom, z.B. durch transkriptomische Daten, schon einige Gene und ihre Sequenz ohne Introns für die zu analysierende Art identifiziert. In dem Fall kann man mit diesen bekannten und validen Vergleichsdaten die HMMs trainieren. Nach dem Training kann man dann die noch unbekannten Gene innerhalb der Genomischen Sequenz mit einer guten Wahrscheinlichkeit identifizieren lassen.
Wirklich problematisch wird es allerdings, wenn man noch keinerlei Gene des Organismus den man untersucht kennt. Denn selbst auf nah verwandte Arten trainierte HMMs bringen hier nur sehr suboptimale Ergebnisse. Ein Trick den man in dem Fall nutzen kann: Man versucht mit den, auf nah Verwandte Organismen trainierten, HMMs einige Gene zu finden um die so gefundenen Gene dann als neues Trainingsset zu benutzen. In der Hoffnung, dass die so trainierten HMMs etwas bessere Ergebnisse liefern als die Ausgangs-Werte. Man sieht aber auf jeden Fall: Die Vorhersage von Genen aus eukaryotischen Genomen ist leider immer noch eine schwierige Aufgabe, erst Recht ohne Referenz-Wissen aus anderen Quellen.
Grafik: Wikipedia, CC-BY-SA 2.0
{kind=link}
Ja gut..
…aber warum müssen’s denn die genomischen Daten sein? Warum tut’s nicht die RNS? Nach was wird da in den Introns gesucht?
Man kann sich seine Daten halt nicht aussuchen. Wenn man auf Gen-Suche gehen will bieten sich sonst natürlich Transkriptome an, eben weil man sich das Introns suchen so bequem
Sparen kann.
Immer noch nicht verstanden
Ist die mRNA nicht zugänglich? Was sind das für Organismen/Proben, und worum geht’s? Systematik, Stammesgeschichte?
Doch ist sie, aber es sind nicht immer alle Gene in einer Zelle angeschaltet und somit liegen nicht immer die mRNAs aller Gene vor. Deswegen muss man, wenn man die komplette genetische Information möchte, auf das Transkriptom bzw. Genom zurückgreifen. Dabei wirft man meistens die Introns raus, da diese oft nicht-codierend und nicht von Interesse sind. Man muss allerdings beachten, dass in Introns die Sequenzen liegen, die für mRNAs codieren. Dies sind kleine regulatorische Moleküle, die Gene regulieren. Von daher darf man die Introns also nicht ganz außer Acht lassen!
@Helmut: Ich meinte vor allem “für mich nicht zugänglich”, weil die Proben keine mRNA mehr hergeben, man kein Geld für eine Transkriptom-Sequenzierung mehr hat oder aus anderen, nicht-biologischen, Gründen. Zumindest ich bin bislang nicht in der Position da so etwas zu fordern. 😉
@Sebastian: Ja, es gibt natürlich auch Gründe warum man das Genom anschauen möchte. Für viele Analysen reicht es aber eben auch sich mRNA oder das Transkriptom anzuschauen, z.B. Über EST-Libraries.
Nicht zu vergessen das EST-Sequenzierung wesentlich billiger als Genomsequenzierung ist, hier ein interessantes Paper das die meisten modernen Sequenzierungsansätze und -methoden vergleicht: link