

Meine Bachelor-Arbeit: Transkriptom-Analysen für jeden?
BLOG: Bierologie

Heutzutage sequenzieren Biologen ja fast alles. Gemeint ist dabei das lesbar machen der genetischen Informationen. Aus den chemischen Bausteinen werden dabei dann lustige Buchstabenfolgen die aus den Anfangsbuchstaben der vier Basen (Adenin (A) oder Guanin (G), Thymin (T) und Cytosin (C)) bestehen die bequem am Computer lesbar sind. Relativ bekannt geworden sind dabei die großen Genom-Sequenzierungs-Projekte. Bei diesen wird die gesamte genetische Information in solche Buchstaben-Ketten verwandelt. Für viele Organismen, darunter auch uns Menschen, gibt es ein solches Referenzgenom. Allerdings ist die Erstellung eines gesamten Genoms trotz moderner Methoden immer noch relativ Zeit- und auch Kosten-Intensiv.
Als Alternative hat sich daher in vielen Bereichen das Erstellen des Transkriptoms anstelle des Genoms durchgesetzt. Bei dem Transkriptom sequenziert man nur jene Teile des Genoms die zu einem Zeitpunkt X gerade auch aktiv benutzt werden um in der Übersetzungsmaschine des Organismus von DNA zu RNA umgeschrieben (transkribiert) werden. Das spart nicht nur Zeit und Geld weil es weniger Material ist was man sequenzieren muss, sondern hat auch ganz praktische Anwendungsfälle: Es gibt nämlich nicht nur ein Transkriptom pro Art sondern ein Transkriptom für einen Zeitpunkt X pro Art. Denn welche Gene gerade aktiv sind verändert sich stark durch verschiedene Dinge: Umweltfaktoren, Lebensalter und eben auch Krankheiten. Die Unterschiede im Transkriptom kann man aktiv ausnutzen: So kann man schauen welche Gene aktiv oder inaktiv werden. Sei es durch Veränderungen in der Umwelt oder auch durch Krankheiten wie Krebs.
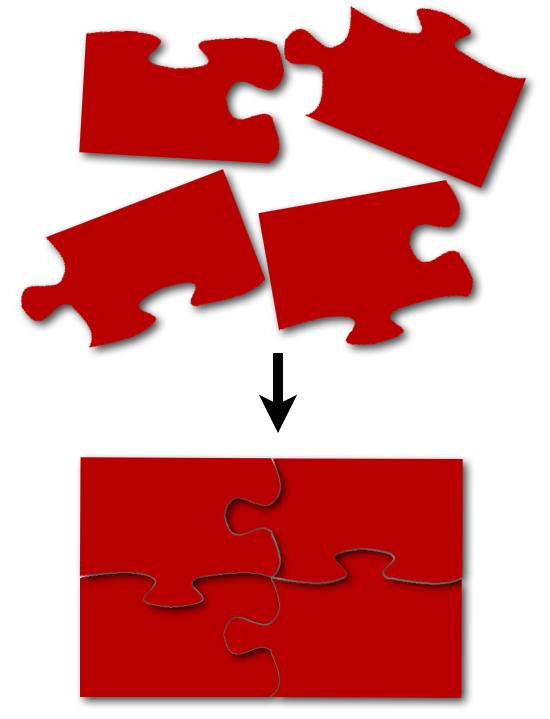
Das klingt nun ganz einfach. Ist aber in der Praxis noch nicht ganz so simpel wie ein Mikrowellengericht zuzubereiten. Denn bei modernen Sequenzier-Techniken bekommt man am Ende nicht eine fertige, lange Zeichenkette mit der man arbeiten kann sondern viele kurze Fragmente. Ähnlich wie Puzzleteile überlappen diese zum Teil dann an den Enden. Was hier mit 4 Teilen noch recht einfach aussieht wird schon komplexer wenn man sich überlegt, dass nicht nur die Anzahl der Teile viel höher ist, sondern auch die Anzahl der Puzzle selbst. Denn am Ende hat man nicht nur ein fertiges Puzzle sondern ganz viele davon. Weil so ein Transkriptom besteht nicht nur aus einem einzelnen Gen sondern aus vielen einzelnen. Als Beispiel: Der Datensatz mit dem ich während meiner Thesis gearbeitet habe bestand aus fast 900.000 Puzzleteilen/Sequenzen die später zu gut 35.000 einzelnen Puzzlen/längeren Sequenzen zusammengesteckt wurden. Das ist schon eine Hausnmmer die man nicht mehr per Hand macht, egal wie sehr man Puzzle-Fan ist. Stattdessen benutzt man dazu sogenannte Assembly-Programme die für einen die Arbeit erledigen.
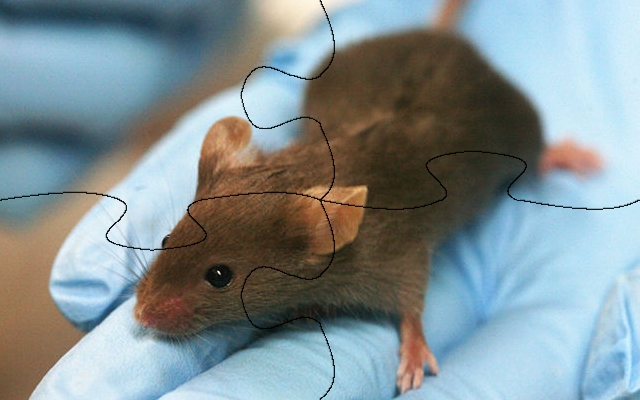
Aber selbst wenn man dann so weit ist, dass man die Puzzle zusammengebaut hat muss man feststellen: Der Informationsgehalt bislang geht gehen Null. Denn die Teile und auch die Puzzles selbst haben alle die gleiche Farbe. Damit man nun erkennt welches Bild eigentlich auf das Puzzle gehört, also was für ein Gen o. Ä. man da gerade hat, muss man wieder ein bisschen Arbeiten: Man muss die Sequenzen annotieren. Dies geschieht üblicherweise über riesige Datenbanken: Man vergleicht seine eigenen Sequenzen gegen andere und schaut ob es diese, oder nahe Verwandte, schon gibt. Aber auch dies ist eine Aufgabe die niemand per Hand machen möchte. Deshalb gibt es auch dafür Software-Lösungen mit passenden Algorithmen die sich um das Suchen kümmern.
Hört sich also so an, als wäre es ein Traum als Biologe mit Sequenzen zu arbeiten: Daten in den Computer, einen Knopf drücken, und ein bisschen später (wobei bisschen bei der Datenmenge meist: Wochen später) die Resultate in der Hand halten. Theoretisch ist das so. In der Praxis funktioniert das nicht so einfach. Alleine die Literatur-Recherche zur Wahl der richtigen Tools im Rahmen meiner Projektarbeit hat gut 8 Wochen gedauert. Und wenn man dann mal die ganzen Paper gelesen hat und sich für ein Set an Programmen entschieden hat fängt der Spaß erst richtig an. Denn Benutzerfreundlichkeit ist in der Open Source-Welt halt noch nicht so richtig angekommen.
Dazu muss man nur mal Gimp und Inkscape mit den überteuerten Äquivalenten von Adobe vergleichen. Wobei die Standard-Programme die man zur Transkriptom-Analyse benötigt lieber gleich auf eine graphische Benutzerführung verzichten und sich auf die Kommandozeile beschränken. So mächtig die Kommandozeile auch ist, so ziemlich alle Studien die ich finden konnte zeigen, dass neue Benutzer damit hoffnungslos überfordert sind. Dazu kommt dann noch die Tatsache, dass so ziemlich jedes Programm was ich mir angeschaut habe den Benutzer mit kryptischen Parametern überschüttet um ihn endgültig in den Wahnsinn zu treiben.
Die meisten Biologen werden mit der Webseite von NCBI BLAST eine Alternative zur Kommadozeilen-Version von BLAST kennen. Mit halbwegs aufgeräumten, graphischem User-Interface und der Möglichkeit die meisten Parameter einfach zu ignorieren. Stattdessen einfach die eigenen Daten hochladen, auf “Run” klicken, und irgendwann die Ergebnisse abholen. Und das hab ich nun, so ähnlich, für die Transkriptom-Analyse nachgebaut. Das Ziel: Seine Sequenzen hochladen, ein paar mal auf “Run” klicken, und am Ende eine hübsche Auswertung der Ergebnisse bekommen. Dabei ist das nicht ganz so trivial. Denn diese Aufgaben, so einfach puzzeln auch klingen mag, sind Rechen- und Zeitintensiv. Deshalb ist es eben nötig die verschiedenen Aufgaben wenn möglich auf verschiedene Computer zu verteilen.
Als Webframework hab ich dabei lustig mit Ruby On Rails rumgespielt während die Last-Verteilung auf verschiedene Maschinen per Distributed Ruby zum Zuge kam. Und im Endeffekt hat das auch ganz gut geklappt. Die Standard-Analysen kann man nun bequem von einem Web-Front-End aus bedienen. Dabei muss man die verschiedenen Programme nun nicht umständlich auf seinem Rechner installieren oder sich per Remote-Zugriff auf Servern einloggen sondern kann bequem die Server-Struktur des Instituts über seinen Browser bedienen. Neben dem initialen Zusammenbauen der Puzzles werden von dem Programm auch Punktmutationen erkannt, genauso wie die Annotation der fertigen Puzzle gegen verschiedene Datenbanken übernommen wird. Wer noch etwas mehr Informationen möchte kann dann auch gleich potentielle Protein-Sequenzen aus den Resultaten erstellen lassen und auch diese gegen Datenbanken annotieren lassen um zu schauen was die Proteine so tun sollten. Getestet habe ich das ganze auch an einem Datensatz und kann nun sagen: Ja, es funktioniert.
Diese simple Klick-Bedienung hat dabei natürlich einen Nachteil: Eine Reduktion der Einstellungsmöglichkeiten macht es leicht möglich die gesamte Software zu bedienen. Unter Umständen fehlen einem damit aber genau die Parameter die man eigentlich haben möchte beziehungsweise benötigt. Bislang fehlt diese Möglichkeit in dem Web-Front-End das ich zusammengeschraubt habe noch. Aber NCBI BLAST löst das Problem dadurch, dass man diese Parameter als “Experten-Einstellungen” freigibt. Und das wäre wohl auch der nächste Schritt den ich implementieren werde. Aber erstmal ist nun eins angesagt, zumindest sobald ich die Verteidigung meiner Arbeit durch hab: Urlaub.
Bild: Wikipedia, CC-BY-SA
{kind=link}
Ist gut, wenn man endlich durch ist, gell. Nun mußt Du Dich erstmal für das Leben danach orientieren. 😉
Findest Du Gimp und Inkscape wirklich benutzerunfreundlich? Ich habe mich mal mit Gimp probiert und dann mit Photoshop und da kam ich mit Photoshop nicht klar. Und für Inkscape gibt es doch auch jede Menge Tutorial, auch eingänglich Videotutorials.
Die Kommandozeile, mit der möchte ich nicht auch anfangen müssen, wenn ich gerade meine Diplomarbeit mache. Aber sonst ist sie eine gute Sache. Ich habe mal eine schöne Analogie gelesen. Eine graphische Oberfläche wie die von M$ ist wie die Chinesische Schrift. Ein Zeichen hat eine Bedeutung. Die Kommandozeile ist wie unsere Sprache. Aus Buchstabenkombinationen wird ein Wort und dann ist dabei noch die Grammatik zu beachten. Wenn man es einigermaßen kann, dann geht vieles mit der Kommandozeile viel schneller.
Weißt Du schon, was Du danach machst? Bioinformatik soll ein gefragtes Fach sein.
Ja, also neu orientieren muss ich mich nun auf jeden Fall. Ich werde auf jeden Fall nicht direkt im Wintersemester mit dem Master weitermachen sondern wohl bis zum Sommersemester 2011 damit warten. Für die Überbrückungszeit suche ich gerade noch eine erfüllende Aufgabe 😉
Zu Gimp & Inkscape: Also ich kenne bislang niemanden der Inkscape wirklich einsteigerfreundlich fand. Das kann aber auch daran liegen, dass ich selbst und auch die Leute die mir ihr Leid geklagt haben von der Adobe-Seite gekommen sind und danach die Alternativen ausprobiert haben. Was ich, relativ neutral betrachtet, bei Gimp z.B. unerträglich finde sind die verschachtelten Menüs wo nichts da ist wo man es erwarten würde.
Zu CLI vs. GUI: Ich arbeit auch viel und gerne in der Shell weil es, ausreichend viel Vorarbeit vorausgesetzt, schneller und effizienter ist. Allerdings muss man eben sehr viel Arbeit vorinvestieren damit man überhaupt so weit kommt. Es gibt ein paar Studien zum Vergleich von CLI & GUI und da ist das Bild fast immer das Gleiche: Wer sich unbedarft an einen Computer setzt der wird mit einer GUI viel schneller zu Ergebnissen kommen als mit einem Terminal.
Und das ist auch so ziemlich das Problem was meine Arbeit lösen sollte: Die meisten Biologen haben weder Zeit noch Lust sich lange mit der Shell und mit Handbüchern der einzelnen Tools herumzuschlagen. Gerade wenn man in dem Bereich nicht viel arbeitet ist die Investition von so viel Zeit ja auch nicht zu rechtfertigen. Dann lieber Quick & Dirty 🙂
Ja, ok, diese verschachtelten Menus und Fenster bei Gimp sind gewöhnungsbedürftig. Aber Phototshop ist auch so ein Monster und nicht intuitiv, da muß man auch wissen, was man macht.
Ja, die Shell lohnt eigentlich nur, wenn man ständig mit ihr zu tun hat. Bei sporadischem Gebrauch vergißt man die Befehle zu leicht und die Einarbeitungszeit ist zu hoch. Und bei einer Diplomarbeit kann die Einarbeitungszeit tödlich sein, weil man keine hat. Aber wenn man richtig zu Potte kommen will, dann lohnt sich die Shell. M$ wollte die Kommandozeile mit win95 eigentlich abschaffen. Und vor ein paar Jahren haben sie dann die powershell eingeführt …
Hochinteressanter Artikel und das Web-frontend finde ich auch einen fantastischen Beitrag zur Open-Source Welt! Respekt.
Am Rande: Ich finde Inkscape eines der intuitivsten Programme, die ich in meiner langen Zeit der Softwareauswahl kennen gelernt habe… (2-3 Videotutorials und los ging es).
Transkriptom-Analysen
Hallo Bastian!
Transkriptom-Analysen sind wirklich eine Fleißarbeit. Es sit einfach ein Wust von Daten der da vor Dir liegt. Ich habe bei meiner Promotion mit SAGE (Serial Analysis of Gene Expression)gearbeitet. Das ich mich etwas abseits vom wissenschaftlichen Mainstream bewegte habe ich immer dann gemerkt wenn andere Wissenschaftler die Nase rümpften und bemerkten: “Warum nicht Mikroarrays?”
Ich hoffe, dass Du vor lauter Buchstaben noch das Gen siehst…;-)
Transkriptomanalyse
Hallo Bastian,
ich bin gerade dabei mich mit Transkriptomanalysen zu beschäftigen. Speziell suche ich einen möglichst preiswerten Anbieter für die Analyse. Ich hab dazu schon einige Internetseiten durchforstet, allerdings bisher noch kein zufriedenstellendes Ergebnis erhalten. Vielleicht kannst du mir ja diesbezüglich weiter helfen, oder hast Adressen an die man sich wenden könnte?
Vielen Dank schon mal im Voraus!
Liebe Grüße Isabelle
@Joe: Ein Kommilitone hat mit SAGE gearbeitet, finde ich auch sehr spannend. Ich glaube aber, auf Dauer kann man vor lauter Buchstaben gar kein Gen mehr sehen, aber das sehen erledigt ja sowieso Software für einen (wenn man gut programmiert hat ;)).
@Isabelle: Sorry, da kann ich dir auch nicht weiterhelfen, da ich die Analysen selbst durchgeführt hab, bin ich auf dem Gebiet der kommerziellen Anbieter überhaupt nicht informiert. Viel Erfolg aber auf jeden Fall damit! 🙂
Hallo Bastian,
mittlerweile hab ich die Daten der Transkriptomanalyse und versuche irgendein Programm zu finden mit dem sich der Berg an Daten bestmöglich auswerten lässt ohne gleich noch ein Informatik Studium anschließen zu müssen. Besteht die Möglichkeit sich deine Bachelor-Arbeit mal anzusehen?